Next-generation sequencing (NGS) libraries from Eclipsebio’s assays typically consist of reads derived from targeted RNA regions. A visualization tool is required to see which genes were present in your samples of interest and, depending on the assay, which specific transcript regions were enriched. Eclipsebio’s service deliverables include BAM, bigWig, and BED files. If you are unfamiliar with working with any of these file types, we describe how to visualize them in this blog.
Interpretation and visualization of NGS data can be challenging, but with genomics viewers such as the Integrative Genomics Viewer (IGV), you can easily explore your data. In this blog, we will walk through how to use the IGV desktop application. To get started, visit the IGV website at https://igv.org/doc/desktop/ and follow the instructions to download the software.
IGV hosts almost 100 different pre-assembled genomes and can support custom genomes in FASTA format. You need to first ensure that you have the proper genome selected. The upper left-hand corner contains a drop-down menu that allows you to switch between different genomes. If you do not see your genome of interest in the drop-down menu, click on the “Genomes” button and choose “Select Hosted Genome” to see all the available genome options.
You next need to import files containing read coverage information from your sequencing data set, called tracks, into the IGV application. To upload tracks, click on the “File” dropdown menu and upload them from your computer with “Load from File” or directly drag and drop the files into IGV. IGV is compatible with many different types of genomic data files, including BAM, bigWig, and BED files. It is important to note that some large data files, such as BAM files, require an associated index file found in the same location on your computer.
When you first open IGV, the default view is completely zoomed out, displaying all the major chromosomes of the reference genome (Figure 1). The top track provides the chromosome locations across the genome reference. Clicking a specific chromosome will navigate to that chromosome. The search bar is for navigation to different regions of the reference genome, and you can enter chromosomes, coordinates (in the format chr:start-stop) or gene names if you are using a hosted genome.
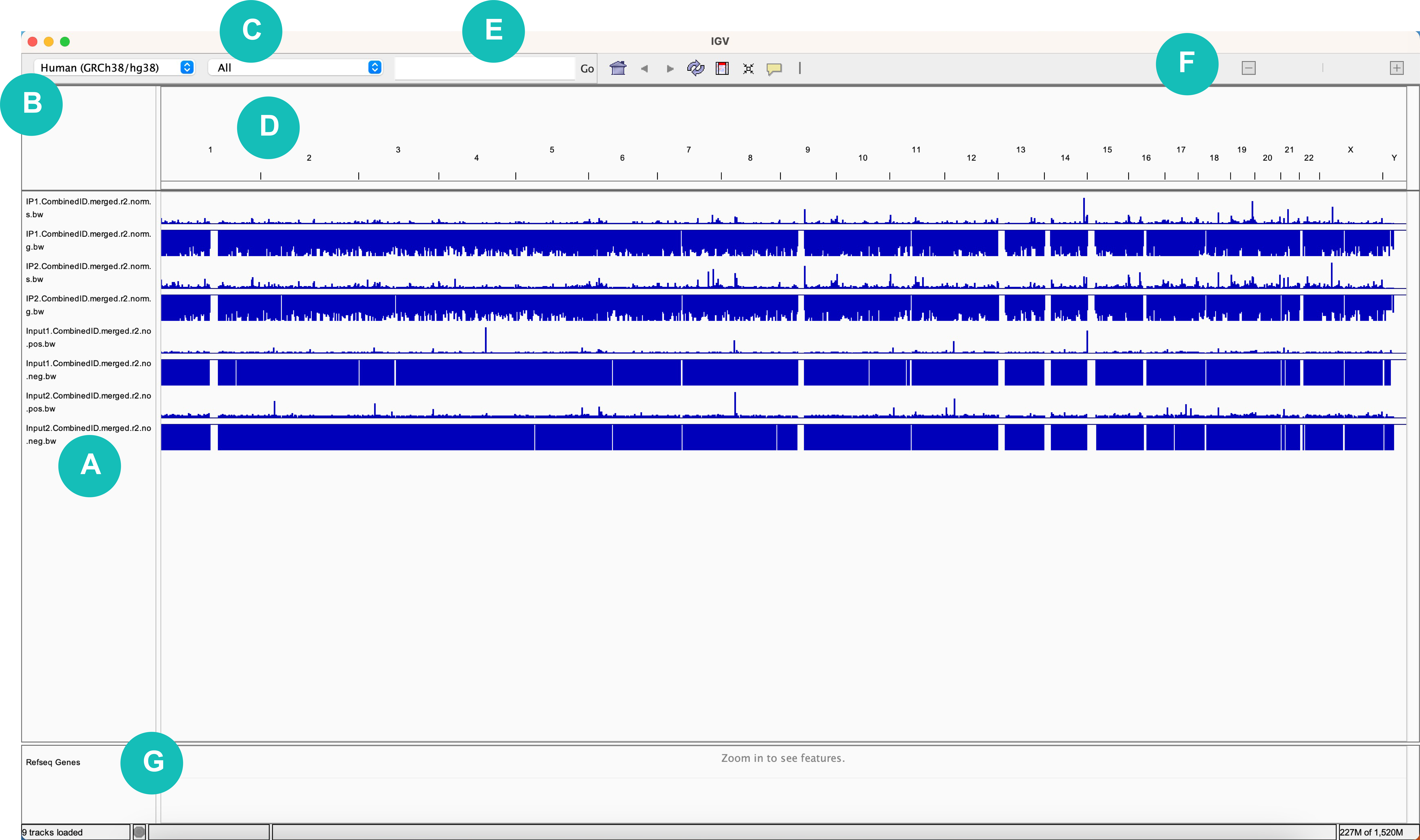
Figure 1: An example IGV window, completely zoomed out. In this image you can see uploaded bigWig tracks (A), the genome dropdown menu with the human hg38 genome assembly selected (B), chromosome dropdown menu (C), chromosome track (D), search bar (E), + and – buttons for zooming in or out (F), and RefSeq gene annotation (G).
Zooming into regions of interest in IGV will show you more detailed data views with some extra tracks such as gene annotations now visible (Figure 2). There are several ways to zoom into a particular region including searching for a specific gene, clicking on a location along the chromosome map, or clicking the + and – buttons in the upper right-hand corner of the window. To move upstream and downstream in the genome browser, click and hold the mouse as you move it left to right.
If you are using a hosted genome, you will see the RefSeq gene annotation preloaded at the bottom of the window. The thickest blue bars represent coding regions of genes, thinner blue bars represent 5’ or 3’ UTRs (untranslated regions), and the thinnest blue lines represent introns. The arrows on the intron dictate the directionality of the gene with > for positive strand genes and < for negative strand genes. By default, the RefSeq annotation is in the “Collapsed” state, meaning all transcripts are collapsed into a single row. Right-click on the RefSeq Genes track and select “Expanded” to see all transcripts annotated at the region.
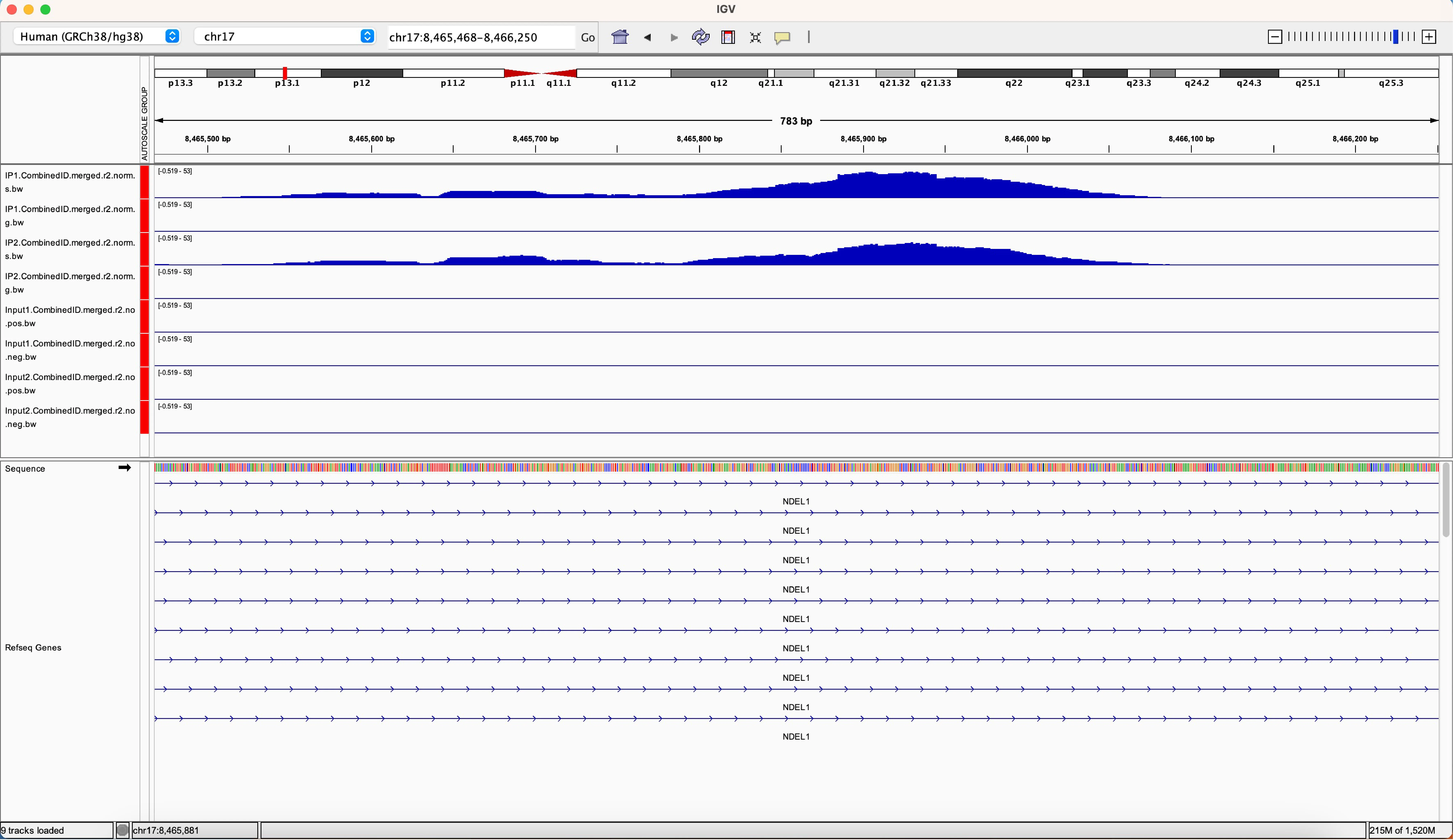
Figure 2: An example IGV window, zoomed into an intronic region of the gene NDEL1. In this example, the RefSeq gene annotation has been expanded to show all transcripts.
You can customize the style of the tracks in multiple ways. You can select and interact with individual tracks by clicking on the name of the track on the left-hand side of the window. You can also select multiple tracks at the same time by holding down the shift or command keys, and then customize a group of tracks together. After selecting your track(s) of interest, right click on the name(s) to see some different options. You can change the color of the tracks, the plotting method, the data range, and more. Note that when tracks are first uploaded, they are not automatically displayed using the same y-axis values. When comparing several sets of tracks, it is helpful to scale them on the same axis using the “Group Autoscale” option. You may also change the display name by selecting one individual track and choosing the “Rename Track” option. Selected tracks can also be removed using the “Remove Track” option.
Once you have styled the tracks to your liking and found an interesting region or gene that you wish to save, you can export your current window as an SVG or PNG image. Select the “File” dropdown menu and choose “Save PNG image” or “Save SVG image.” You can then use these images to create figures for publication.
With Eclipsebio’s RBP-eCLIP service, we have made viewing your data even easier. Our data analysis platform, Lunar, includes the automatic upload of tracks into a web browser version of IGV. The window will be preloaded with bigWig tracks for each sample, clusters and peaks for each immunoprecipitation (IP) sample, and the specific reference and annotation files used by Eclipsebio’s analysis pipeline to save you the time and effort needed to create your own IGV session (Figure 3). HTML reports provide a link to each peak in IGV, allowing you to easily jump to a binding site or gene of interest.
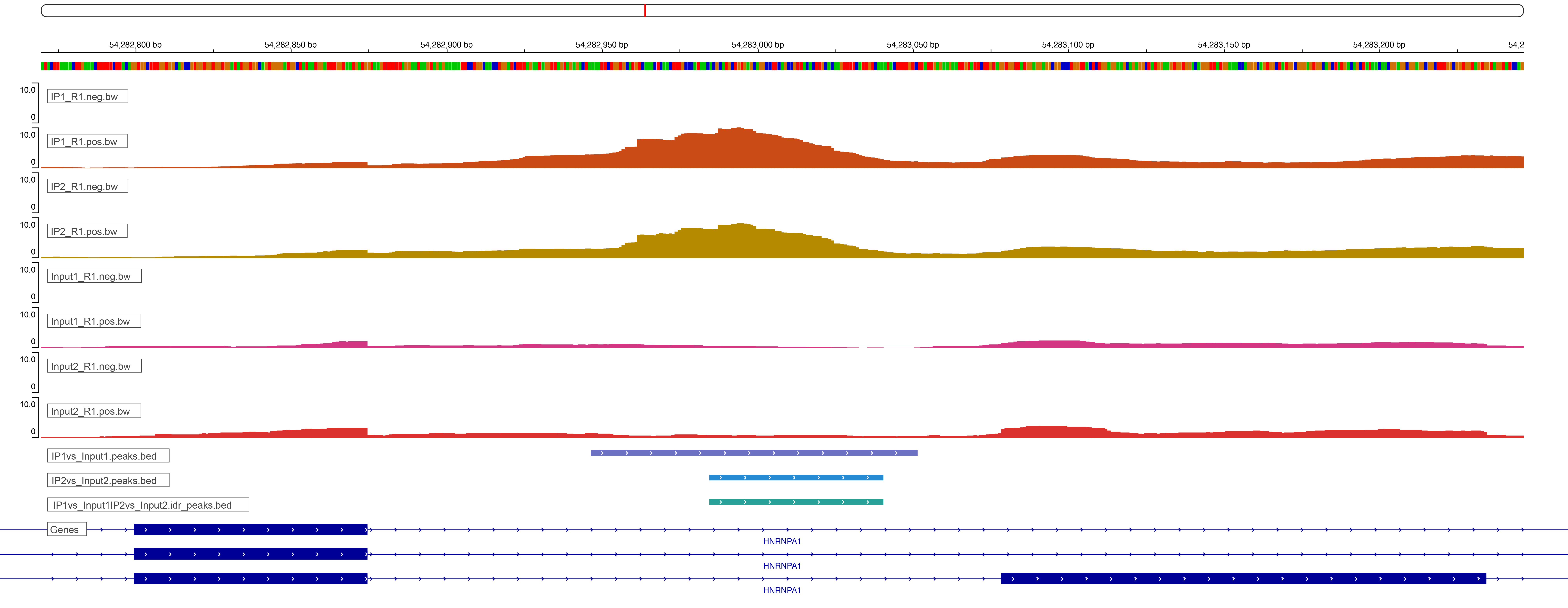
Figure 3: An example IGV window from Eclipsebio’s Lunar data analysis platform. A link within the HTML report allowed us to jump directly to this region in IGV.
Aside from what we have detailed above, IGV has many other capabilities for interacting with and visualizing NGS data. See the IGV website to download the IGV application and to view more complete documentation.
Latest eBlogs
Charting a New Era: How RNA Is Unlocking N‑of‑1 Cures
RNA is unlocking the ability to develop life‑saving therapies at unprecedented speed. The recent success with baby KJ demonstrates how RNA medicine can move a personalized treatment from diagnosis to clinic in less than a year.
The three pillars of AI in RNA biology: why data is the hardest to get right
Artificial intelligence (AI) is transforming how we approach RNA research and drug discovery. In this eBlog we review how data is one of the key pillars for the successful use of AI.